
ACALABRUTINIB 100 mg, gélules, boîte de 7 plaquettes de 8
Retiré du marché le : 26/11/2020
Dernière révision : 09/07/2020
Taux de TVA : 0%
Laboratoire exploitant : ASTRAZENECA
Source :

Acalabrutinib est indiqué chez des patients adultes atteints d'une leucémie lymphoïde chronique (LLC) :
• en monothérapie ou en association à l'obinutuzumab, chez les patients non précédemment traités, sans délétion 17p ni mutation du gène TP53 et inéligibles à un traitement à base de fludarabine à pleine dose,
• en monothérapie chez les patients intolérants ou inéligibles à un traitement par ibrutinib.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition.
Hémorragies
Des événements hémorragiques majeurs, y compris des hémorragies au niveau du système nerveux central et au niveau gastro-intestinale, certaines d'évolution fatale, sont survenus chez des patients atteints de cancers hématologiques et traités par acalabrutinib en monothérapie et en association avec l'obinutuzumab. Ces événements se sont produits chez des patients avec et sans thrombopénie. Dans l'ensemble, les événements hémorragiques étaient des événements moins sévères, notamment des hématomes et des pétéchies (voir rubrique Effets indésirables).
Les patients recevant des antithrombotiques peuvent présenter un risque accru d'hémorragie. Utiliser les antithrombotiques avec précaution et envisager une surveillance supplémentaire à la recherche de signes d'hémorragie si une utilisation concomitante est médicalement nécessaire.
Évaluer le rapport bénéfice/risque d'une suspension d'acalabrutinib pendant au moins 3 jours avant et après une intervention chirurgicale.
Infections
Des infections (bactériennes, virales ou fongiques) graves, y compris des événements fatals, sont survenues chez des patients atteints de cancers hématologiques et traités par acalabrutinib en monothérapie et en association avec l'obinutuzumab. Des cas d'infections dues à une réactivation du virus de l'hépatite B (VHB) et du virus varicelle-zona (VZV), d'aspergillose et de leucoencéphalopathie multifocale progressive (LEMP) sont survenus (voir rubrique Effets indésirables).
Des cas de réactivation du virus de l'hépatite B (VHB) ont été rapportés chez des patients recevant acalabrutinib. Le statut vis-à-vis du VHB doit être établi avant de commencer le traitement par acalabrutinib. En cas de sérologie positive pour l'hépatite B, un hépatologue doit être consulté avant le début du traitement et le patient doit être surveillé et pris en charge conformément à la pratique médicale en vigueur afin de prévenir une réactivation de l'hépatite B.
Des cas de leucoencéphalopathie multifocale progressive (LEMP), y compris des cas d'évolution fatale, ont été rapportés après l'utilisation d'acalabrutinib dans le contexte d'un traitement immunosuppresseur antérieur ou concomitant. Les prescripteurs doivent envisager la LEMP comme diagnostic différentiel en cas d'apparition ou d'aggravation de signes ou de symptômes neurologiques, cognitifs ou comportementaux. Si une LEMP est suspectée, les évaluations diagnostiques appropriées doivent être réalisées et le traitement par acalabrutinib doit être suspendu jusqu'à l'exclusion du diagnostic de LEMP. En cas de doute, il convient d'envisager d'adresser le patient à un neurologue et de prendre les mesures diagnostiques appropriées, à savoir notamment une IRM de préférence avec administration d'un produit de contraste, une analyse du liquide céphalo-rachidien (LCR) à la recherche d'ADN du virus JC et de nouvelles évaluations neurologiques.
Envisager une prophylaxie conformément à la pratique habituelle chez les patients qui présentent un risque accru d'infections opportunistes. Surveiller les patients à la recherche de signes et de symptômes d'infection et traiter lorsque cela est médicalement approprié.
Cytopénies
Des cytopénies de grade 3 ou 4 apparues sous traitement, y compris des cas de neutropénie, d'anémie et de thrombopénie, sont survenues chez des patients atteints de cancers hématologiques et traités par acalabrutinib en monothérapie et en association avec l'obinutuzumab. Surveiller la numération formule sanguine lorsque cela est médicalement indiqué (voir rubrique Effets indésirables).
Seconds cancers primitifs
Des seconds cancers primitifs, y compris des cancers non cutanés, sont survenus chez des patients atteints de cancers hématologiques et traités par acalabrutinib en monothérapie et en association avec l'obinutuzumab. Des cancers cutanés ont été fréquemment rapportés. Surveiller les patients à la recherche de cancers cutanés et leur conseiller de se protéger du soleil (voir rubrique Effets indésirables).
Fibrillation auriculaire
Des cas de fibrillation auriculaire / flutter sont survenus chez des patients atteints de cancers hématologiques et traités par acalabrutinib en monothérapie et en association avec l'obinutuzumab. Surveiller les patients à la recherche de symptômes (p. ex. palpitations, étourdissements, syncope, douleurs thoraciques, dyspnée) de fibrillation auriculaire et de flutter et réaliser un ECG lorsque cela est médicalement indiqué (voir rubriques Interactions avec d'autres médicaments et autres formes d'interactions et Posologie et mode d'administration). Chez les patients qui développent une fibrillation auriculaire sous traitement par acalabrutinib, une évaluation approfondie du risque de maladie thrombo-embolique doit être réalisée. Chez les patients qui présentent un risque élevé de maladie thrombo-embolique, un traitement étroitement contrôlé par anticoagulants et des options thérapeutiques alternatives à acalabrutinib doivent être envisagés.
Autres médicaments
La co-administration d'inhibiteurs puissants du CYP3A avec l'acalabrutinib peut entraîner une augmentation de l'exposition à l'acalabrutinib et donc un risque accru de toxicité. Réciproquement, la co-administration d'inducteurs du CYP3A peut entraîner une diminution de l'exposition à l'acalabrutinib et donc un risque de manque d'efficacité. Par conséquent, des traitements alternatifs aux inhibiteurs ou inducteurs puissants du CYP3A doivent être envisagés si possible. Les patients doivent être surveillés attentivement à la recherche d'éventuels signes de toxicité si un inhibiteur puissant du CYP3A doit être utilisé (voir rubriques Posologie et mode d'administration et Interactions avec d'autres médicaments et autres formes d'interactions). Si un inducteur puissant du CYP3A doit être utilisé, augmenter la dose d'acalabrutinib à 200 mg deux fois par jour.
Acalabrutinib contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Sur les 1 263 patients traités par acalabrutinib, les effets indésirables de tout grade les plus fréquents rapportés chez les patients recevant l'acalabrutinib étaient les suivants : infection (68 %), céphalées (38,7 %), diarrhée (38 %), hématomes (34,9 %), douleurs musculo-squelettiques (35,2 %), nausées
(22,6 %), fatigue (23 %) et rash (22,2 %).
Les effets indésirables de grade ≥ 3 fréquemment rapportés étaient les suivants : infection (18,3 %), neutropénie (17 %), anémie (7,4 %) thrombopénie (5,5 %), second cancer primitif (4,1 %), hémorragie/hématome (1,7 %) et fibrillation auriculaire (1,2 %).
Tableau récapitulatif des effets indésirables
Les effets indésirables suivants ont été identifiés dans les études cliniques conduites chez des patients recevant acalabrutinib pour le traitement de cancers hématologiques. La durée médiane du traitement par acalabrutinib dans la population poolée était de 26,2 mois.
Les effets indésirables sont présentés par classe de systèmes d'organes (SOC) MedDRA. Au sein de chaque classe de systèmes d'organes, les effets indésirables sont classés par fréquence, les effets indésirables les plus fréquents figurant en premier. De plus, la catégorie de fréquence correspondant à chaque effet indésirable est définie de la manière suivante : très fréquent (≥1/10) ; fréquent (≥1/100 à <1/10) ; peu fréquent (≥1/1 000 à <1/100) ; rare (≥1/10 000 à <1/1 000) ; très rare (<1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 3. Effets indésirables* des patients atteints de cancers hématologiques et traités par
acalabrutinib (n = 1 263)
| SOC MedDRA | Terme MedDRA | Descripteur CIOMS/ Fréquence globale (tous grades CTCAE) | Fréquence des événements de grade CTCAE ≥ 3† |
| Affections hématologiques et du système lymphatique | Neutropénie | Très fréquent (18,5 %) | 17 % |
| Anémie | Très fréquent (13,5 %) | 7,4 % | |
| Thrombopénie | Fréquent (9,8 %) | 5,5 % | |
| Lymphocytose | Peu fréquent (0,3 %) | 0,2 % | |
| Affections cardiaques | Fibrillation auriculaire/flutter † | Fréquent (4,2 %) | 1,2 % |
| Affections du système nerveux | Céphalées | Très fréquent (38,7 %) | 1 % |
| Étourdissements | Très fréquent (15,2 %) | 0,2 % | |
| Affections gastro- intestinales | Diarrhée | Très fréquent (38 %) | 2,9 % |
| Nausées | Très fréquent (22,6 %) | 1 % | |
| Constipation | Très fréquent (15,5 %) | 0,1 % | |
| Vomissements | Très fréquent (14,3 %) | 0,9 % | |
| Douleurs abdominales† | Très fréquent (12,9 %) | 1 % | |
| Troubles généraux et anomalies au site d'administration | Fatigue | Très fréquent (23 %) | 1,7 % |
| Asthénie | Fréquent (5,7 %) | 0,7 % | |
| Troubles du métabolisme et de la nutrition | Syndrome de lyse tumorale± | Peu fréquent (0,7 %) | 0,6 % |
| Affections musculo- squelettiques et du tissu conjonctif | Douleurs musculo- squelettiques† | Très fréquent (35,2 %) | 1,7 % |
| Arthralgie | Très fréquent (20,5 %) | 0,8 % | |
| Infections et Infestations | Infection des voies aériennes supérieures | Très fréquent (23,7 %) | 1 % |
| Pneumopathie | Fréquent (9 %) | 5,1 % | |
| Rhinopharyngite | Fréquent (8,5 %) | 0,1 % | |
| Bronchite | Fréquent (6,6 %) | 0,3 % | |
| Infections à herpesvirus† | Fréquent (6 %) | 0,8 % | |
| Infections à Aspergillus† | Peu fréquent (0,4 %) | 0,3 % | |
| Réactivation de l'hépatite B | Peu fréquent (0,2 %) | 0,1 % | |
| Sinusite | Peu fréquent (0,1 %) | 0 % | |
| Infection des voies urinaires | Peu fréquent (0,1 %) | 0 % | |
| Leucoencéphalopathie multifocale progressive | Peu fréquent (0,1 %) | 0,1 % | |
| Tumeurs bénignes, malignes et non précisées | Second cancer primitif† | Très fréquent (12,4 %) | 4,1 % |
| Cancer cutané non mélanocytaire† | Fréquent (6,8 %) | 0,5 % | |
| SCP sauf cancer cutané non mélanocytaire† | Fréquent (6,5 %) | 3,7 % | |
| Affections de la peau et du tissu sous- cutané | Éruption cutanée† | Très fréquent (22,2 %) | 0,8 % |
| Affections vasculaires | Hématomes† | Très fréquent (34,9 %) | - |
| Ecchymoses | Fréquent (8,7 %) | 0 % | |
| Pétéchies | Fréquent (7,7 %) | 0 % | |
| Contusion | Rare (0 %) | 0 % | |
| Hémorragie† | Très fréquent (13,5 %) | 1,7 % | |
| Hémorragie gastro- intestinale | Fréquent (2,5 %) | 0,6 % | |
| Hémorragie intracrânienne | Fréquent (1 %) | 0,4 % | |
| Épistaxis | Fréquent (7,3 %) | 0,2 % |
*D'après la version 4.03 de la classification NCI CTCAE (National Cancer Institute Common Terminology
Criteria for Adverse Events).
†Inclut de multiples termes d'effet indésirable.
±Un cas de syndrome de lyse tumorale induit par le médicament a été observé dans le bras acalabrutinib dans l'étude ASCEND.
Description d'effets indésirables particuliers
Arrêts de traitement et réductions de dose en raison d'effets indésirables
Sur les 1 263 patients traités par Calquence, des arrêts de traitement en raison d'événements indésirables ont été rapportés chez 9,6 % des patients. Il s'agissait notamment de cas de pneumopathie, de thrombopénie et de diarrhée. Des réductions de dose en raison d'événements indésirables ont été rapportées chez 4,7 % des patients.
| SOC MedDRA | Terme MedDRA | Descripteur CIOMS/ Fréquence globale (tous grades CTCAE) | Fréquence des événements de grade CTCAE ≥ 3† |
| Affections hématologiques et du système lymphatique | Neutropénie | Très fréquent (18,5 %) | 17 % |
| Anémie | Très fréquent (13,5 %) | 7,4 % | |
| Thrombopénie | Fréquent (9,8 %) | 5,5 % | |
| Lymphocytose | Peu fréquent (0,3 %) | 0,2 % | |
| Affections cardiaques | Fibrillation auriculaire/flutter † | Fréquent (4,2 %) | 1,2 % |
| Affections du système nerveux | Céphalées | Très fréquent (38,7 %) | 1 % |
| Étourdissements | Très fréquent (15,2 %) | 0,2 % | |
| Affections gastro- intestinales | Diarrhée | Très fréquent (38 %) | 2,9 % |
| Nausées | Très fréquent (22,6 %) | 1 % | |
| Constipation | Très fréquent (15,5 %) | 0,1 % | |
| Vomissements | Très fréquent (14,3 %) | 0,9 % | |
| Douleurs abdominales† | Très fréquent (12,9 %) | 1 % | |
| Troubles généraux et anomalies au site d'administration | Fatigue | Très fréquent (23 %) | 1,7 % |
| Asthénie | Fréquent (5,7 %) | 0,7 % | |
| Troubles du métabolisme et de la nutrition | Syndrome de lyse tumorale± | Peu fréquent (0,7 %) | 0,6 % |
| Affections musculo- squelettiques et du tissu conjonctif | Douleurs musculo- squelettiques† | Très fréquent (35,2 %) | 1,7 % |
| Arthralgie | Très fréquent (20,5 %) | 0,8 % | |
| Infections et Infestations | Infection des voies aériennes supérieures | Très fréquent (23,7 %) | 1 % |
| Pneumopathie | Fréquent (9 %) | 5,1 % | |
| Rhinopharyngite | Fréquent (8,5 %) | 0,1 % | |
| Bronchite | Fréquent (6,6 %) | 0,3 % | |
| Infections à herpesvirus† | Fréquent (6 %) | 0,8 % | |
| Infections à Aspergillus† | Peu fréquent (0,4 %) | 0,3 % | |
| Réactivation de l'hépatite B | Peu fréquent (0,2 %) | 0,1 % | |
| Sinusite | Peu fréquent (0,1 %) | 0 % | |
| Infection des voies urinaires | Peu fréquent (0,1 %) | 0 % | |
| Leucoencéphalopathie multifocale progressive | Peu fréquent (0,1 %) | 0,1 % | |
| Tumeurs bénignes, malignes et non précisées | Second cancer primitif† | Très fréquent (12,4 %) | 4,1 % |
| Cancer cutané non mélanocytaire† | Fréquent (6,8 %) | 0,5 % |
| SCP sauf cancer cutané non mélanocytaire† | Fréquent (6,5 %) | 3,7 % | |
| Affections de la peau et du tissu sous- cutané | Éruption cutanée† | Très fréquent (22,2 %) | 0,8 % |
| Affections vasculaires | Hématomes† | Très fréquent (34,9 %) | - |
| Ecchymoses | Fréquent (8,7 %) | 0 % | |
| Pétéchies | Fréquent (7,7 %) | 0 % | |
| Contusion | Rare (0 %) | 0 % | |
| Hémorragie† | Très fréquent (13,5 %) | 1,7 % | |
| Hémorragie gastro- intestinale | Fréquent (2,5 %) | 0,6 % | |
| Hémorragie intracrânienne | Fréquent (1 %) | 0,4 % | |
| Épistaxis | Fréquent (7,3 %) | 0,2 % |
*D'après la version 4.03 de la classification NCI CTCAE (National Cancer Institute Common Terminology
Criteria for Adverse Events).
†Inclut de multiples termes d'effet indésirable.
±Un cas de syndrome de lyse tumorale induit par le médicament a été observé dans le bras acalabrutinib dans l'étude ASCEND.
Description d'effets indésirables particuliers
Arrêts de traitement et réductions de dose en raison d'effets indésirables
Sur les 1 263 patients traités par acalabrutinib, des arrêts de traitement en raison d'événements indésirables ont été rapportés chez 9,6 % des patients. Il s'agissait notamment de cas de pneumopathie , de thrombopénie et de diarrhée. Des réductions de dose en raison d'événements indésirables ont été rapportées chez 4,7 % des patients.
Tableau 4. Anomalies biologiques hématologiques apparues sous traitement pour l'acalabrutinib (n = 1 263)
| SOC MedDRA | Terme MedDRA | Descripteur CIOMS/ Fréquence globale (tous grades CTCAE) | Fréquence des événements de grade CTCAE 3- 4 |
| Investigations (conclusions basées sur les résultats des tests présentés comme des changements de grade CTCAE) | Diminution du nombre absolu de neutrophiles | Très fréquent (44,6 %) | 23,2 % |
| Diminution de l'hémoglobine | Très fréquent (42,8 %) | 9,9 % | |
| Diminution des plaquettes | Très fréquent (33,7 %) | 7,6 % |
D'après la version 4.03 de la classification NCI CTCAE (National Cancer Institute Common Terminology Criteria for Adverse Events).
Description d'effets indésirables particuliers
Sujets âgés
Sur les 1 263 patients des essais cliniques sur acalabrutinib en monothérapie, 42 % étaient âgés de plus de 65 ans et de moins de 75 ans et 23 % avaient 75 ans ou plus. Aucune différence cliniquement pertinente au niveau de la sécurité ou de l'efficacité n'a été observée entre les patients âgés de < 65 ans et ceux âgés de ≥ 65 ans.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté à l'aide de la fiche de déclaration des effets indésirables et de signalement de grossesse disponible dans le Protocole d'utilisation thérapeutique et de recueil d'informations (voir Annexes B4 et B5 du PUT).
SURVEILLANCE DU TRAITEMENT :
- Recherche de signes et symptômes d'infection.
- NFS lorsque cela est médicalement indiqué.
- Recherche de cancers cutanés.
- Recherche de symptômes (p. ex. palpitations, étourdissements,
syncope, douleurs thoraciques, dyspnée) de fibrillation auriculaire et
de flutter et réaliser un ECG lorsque cela
est médicalement indiqué.
CONSULTER IMMEDIATEMENT UN MEDECIN OU ALLER AU SERVICE D'URGENCES LE PLUS PROCHE en cas de :
- Selles noires ou contenant du sang, urines de couleur rose ou marron, saignements de nez,
bleus, saignement inexpliqué, vomissements ou crachats de sang, étourdissements, faiblesse,
confusion, troubles de la parole, ou maux de tête persistants (signes de problèmes de
saignement, également désignés par le terme « hémorragie »).
- Infections (bactériennes, virales ou à champignons), se manifestant notamment par les signes et
symptômes suivants : fièvre ou frissons ou syndrome pseudo-grippal.
- Battements cardiaques rapides ou anormaux, étourdissements, sensation d'être sur le point de
s'évanouir, gêne thoracique ou essoufflement (signes de troubles du rythme cardiaque, également
désignés par les termes « fibrillation auriculaire /flutter »).
UTILISER UNE PROTECTION SOLAIRE en cas d'exposition à la lumière du soleil.
NE PAS ALLAITER pendant le traitement et pendant 2 semaines après la dernière dose.
PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (étourdissements, faiblesse, fatigue).
PREVENIR le médecin de la prise de ce médicament en cas d'intervention chirurgicale programmée.
EVITER de consommer des préparations à base
de plantes contenant du millepertuis (Hypericum perforatum) qui
pourrait diminuer de manière imprévisible les concentrations
plasmatiques du médicament.
Femmes en âge de procréer/contraception chez les femmes
Il doit être conseillé aux femmes en âge de procréer de ne pas tomber enceintes pendant le traitement par acalabrutinib.
Grossesse
Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la reproduction (voir rubrique Données de sécurité précliniques.). L'administration d'acalabrutinib à des lapines gravides à des expositions correspondant à 4 fois l'ASC chez l'être humain à la dose recommandée a été associée à une réduction de la croissance foetale. Une dystocie a été observée dans une étude chez le rat impliquant une administration à partir de l'implantation et pendant la gestation, la parturition et l'allaitement à des expositions correspondant à > 2,3 fois l'ASC chez l'être humain à la dose recommandée (voir rubrique Données de sécurité précliniques). Les données cliniques sur l'utilisation d'acalabrutinib chez la femme enceinte sont insuffisantes pour mettre en évidence un risque de malformation congénitale majeure et de fausse couche en lien avec le médicament. À titre de précaution, il est préférable d'éviter l'utilisation d'acalabrutinib pendant la grossesse.
Allaitement
On ne sait pas si l'acalabrutinib est excrété dans le lait maternel. Il n'existe pas de données sur l'effet de l'acalabrutinib sur l'enfant allaité ou sur la production de lait. L'acalabrutinib et son métabolite actif étaient présents dans le lait de rates allaitantes. Un risque pour l'enfant allaité ne peut être exclu. Il est conseillé aux mères de ne pas allaiter pendant le traitement par acalabrutinib et pendant 2 semaines après la dernière dose.
Fertilité
Il n'existe pas de données de l'effet d'acalabrutinib sur la fertilité humaine. Dans une étude non clinique conduite sur l'acalabrutinib chez des rats mâles et femelles, aucun effet indésirable n'a été observé sur les paramètres de la fertilité (voir rubrique Données de sécurité précliniques).
L’acalabrutinib est principalement métabolisé par l’enzyme 3A4 du cytochrome P450 (CYP3A4).
Substances actives pouvant augmenter les concentrations plasmatiques d’acalabrutinib
Inhibiteurs du CYP3A, de la P-gp et/ou de la BCRP
La co-administration avec un inhibiteur puissant du CYP3A, de la P-gp et/ou de la BCRP (200 mg d'itraconazole une fois par jour pendant 5 jours) a multiplié la Cmax et l'aire sous la courbe (ASC) de l'acalabrutinib respectivement par 3,9 et 5,0 chez des sujets sains (N = 17).
Envisager des traitements alternatifs qui n'inhibent pas fortement l'activité du CYP3A, de la P-gp et/ou de la BCRP. Sinon, si les inhibiteurs puissants du CYP3A, de la P-gp et/ou de la BCRP (p. ex. kétoconazole, conivaptan, clarithromycine, indinavir, itraconazole, ritonavir, télaprévir, posaconazole, voriconazole) doivent être utilisés à court terme, interrompre acalabrutinib (voir rubrique Posologie et mode d'administration).
Les simulations à l'aide d'un modèle pharmacocinétique physiologique (PBPK) réalisées avec l'acalabrutinib et des inhibiteurs modérés du CYP3A (érythromycine, fluconazole, diltiazem) ont montré que la co-administration de ces médicaments multipliait la Cmax et l'ASC de l'acalabrutinib par environ 2 à 3. Lorsque l'acalabrutinib est co-administré avec des inhibiteurs modérés du CYP3A, réduire la dose d'acalabrutinib à 100 mg une fois par jour.
Substances actives pouvant diminuer les concentrations plasmatiques d'acalabrutinib
Inducteurs du CYP3A
La co-administration avec un inducteur puissant du CYP3A (600 mg de rifampicine une fois par jour pendant 9 jours) a diminué la Cmax et l'ASC de l'acalabrutinib respectivement de 68 % et 77 % chez des sujets sains (N = 24).
Envisager des traitements alternatifs aux inducteurs puissants de l'activité du CYP3A (p. ex. phénytoïne, rifampicine, carbamazépine). Éviter le millepertuis qui pourrait diminuer de manière imprévisible les concentrations plasmatiques d'acalabrutinib (voir rubrique Posologie et mode d'administration). Si un inducteur puissant du CYP3A ne peut pas être évité, augmenter la dose d'acalabrutinib à 200 mg deux fois par jour (voir rubrique Posologie et mode d'administration).
Médicaments diminuant l'acidité gastrique
La solubilité de l'acalabrutinib diminue avec l'augmentation du pH. La co-administration de l'acalabrutinib avec un antiacide (1 g de carbonate de calcium) a diminué l'ASC de l'acalabrutinib de 53 % chez des sujets sains. La co-administration avec un inhibiteur de la pompe à protons (40 mg d'oméprazole pendant 5 jours) a diminué l'ASC de l'acalabrutinib de 43 %.
Si un traitement par un médicament diminuant l'acidité gastrique est nécessaire, envisager l'utilisation d'un antiacide (p. ex. carbonate de calcium) ou d'un antagoniste des récepteurs H2 (p. ex. ranitidine ou famotidine). Pour une utilisation avec des antiacides, les prises des deux médicaments doivent être espacées d'au moins 2 heures (voir rubrique Posologie et mode d'administration). Pour une utilisation avec des antagonistes des récepteurs H2, prendre acalabrutinib 2 heures avant (ou 10 heures après) la prise de l'antagoniste des récepteurs H2.
En raison de la longue durée d'action des inhibiteurs de la pompe à protons, l'intervalle entre leur administration et celle d'acalabrutinib pourrait ne pas être suffisant pour empêcher l'interaction des deux médicaments (voir rubrique Posologie et mode d'administration).
Substances actives dont les concentrations plasmatiques peuvent être altérées par acalabrutinib Substrats du CYP3A
D'après les données in vitro et la modélisation PBPK, aucune interaction avec les substrats du CYP n'est attendue aux concentrations cliniquement pertinentes (voir rubrique Propriétés pharmacocinétiques).
Effet de l'acalabrutinib sur les substrats du CYP1A2
Les études in vitro indiquent que l'acalabrutinib induit le CYP1A2. La co-administration d'acalabrutinib avec des substrats du CYP1A2 (p. ex. théophylline, caféine) peut diminuer l'exposition à ces substances.
Effets de l'acalabrutinib et de son métabolite actif, l'ACP-5862, sur les systèmes de transport médicamenteux
L'acalabrutinib peut augmenter l'exposition aux substrats de la BCRP co-administrés (p. ex. méthotrexate) par inhibition de la BCRP intestinale (voir rubrique Propriétés pharmacocinétiques). Pour minimiser le risque d'interaction au niveau du tractus gastro-intestinal, les substrats de la BCRP administrés par voie orale et à marge thérapeutique étroite tels que le méthotrexate doivent être pris au moins 6 heures avant ou après l'acalabrutinib.
L'ACP-5862 peut augmenter l'exposition aux substrats de la MATE1 co-administrés (p. ex. metformine) par inhibition de la MATE1 (voir rubrique Propriétés pharmacocinétiques). ). Les patients prenant des médicaments concomitants dont l'élimination dépend de la MATE1 (p. ex. metformine) doivent être surveillés à la recherche de signes de modification de la tolérance en raison d'une exposition accrue au médicament concomitant pendant le traitement par acalabrutinib.
Le traitement par ce médicament doit être initié et supervisé par un médecin expérimenté dans l'utilisation des médicaments anticancéreux.
Posologie
La dose recommandée est de 100 mg d'acalabrutinib deux fois par jour (soit une dose quotidienne totale de 200 mg). Voir le Résumé des Caractéristiques du Produit de l'obinutuzumab pour les informations relatives à la posologie recommandée de ce médicament.
L'intervalle entre deux prises est d'environ 12 heures.
Le traitement par acalabrutinib doit être poursuivi jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
Ajustements posologiques
Effets indésirables
Les modifications posologiques recommandées concernant acalabrutinib suite à des effets indésirables de grade ≥3 sont présentées dans le Tableau 1.
Tableau 1. Ajustements posologiques recommandés en cas d'effets indésirables*
| Effet indésirable | Survenue de l'effet indésirable | Modification posologique (Dose de départ = 100 mg environ toutes les 12 heures) |
| Thrombopénie de grade 3 associée à des saignements, Thrombopénie de grade 4 | Première et deuxième fois | Interrompre acalabrutinib Une fois la toxicité revenue au grade 1 ou au niveau initial, acalabrutinib peut être repris à 100 mg environ toutes les 12 heures |
| | Interrompre acalabrutinib | |
| Ou Neutropénie de grade 4 durant plus de 7 jours | Troisième fois | Une fois la toxicité revenue au grade 1 ou au niveau initial, acalabrutinib peut être repris à une fréquence réduite de 100 mg une fois par jour |
| | Arrêter acalabrutinib | |
| Toxicités non hématologiques de grade 3 ou plus | Quatrième fois | |
*Gradation des effets indésirables établie d'après la version 4.03 de la classification NCI CTCAE (National Cancer Institute Common Terminology Criteria for Adverse Events).
Interactions médicamenteuses
L'utilisation d'acalabrutinib avec des inhibiteurs ou des inducteurs du CYP3A et des médicaments diminuant l'acidité gastrique est présentée dans le Tableau 2 (voir rubrique Posologie et mode d'administration).
Tableau 2. Utilisation avec des inhibiteurs ou des inducteurs du CYP3A et des médicaments diminuant l'acidité gastrique
| | Médicament co- administré | Utilisation recommandée d'acalabrutinib |
| Inhibiteurs du CYP3A | Inhibiteur puissant du CYP3A | Éviter l'utilisation concomitante. Si ces inhibiteurs doivent être utilisés à court terme (comme des anti-infectieux sur une durée maximale de sept jours), interrompre acalabrutinib |
| Inhibiteur modéré du CYP3A | 100 mg une fois par jour. | |
| Inhibiteur faible du CYP3A | Pas d'ajustement posologique. | |
| Inducteurs du CYP3A | Inducteur puissant du CYP3A | Éviter l'utilisation concomitante. Si ces inducteurs ne peuvent pas être évités, augmenter la dose d'acalabrutinib à 200 mg environ toutes les 12 heures. |
| Médicaments diminuant l'acidité gastrique | Inhibiteurs de la pompe à protons | Éviter l'utilisation concomitante. |
| Antagonistes des récepteurs H2 | Prendre acalabrutinib 2 heures avant (ou 10 heures après) la prise d'un antagoniste des récepteurs H2. | |
| Antiacides | Les prises des deux médicaments doivent être espacées d'au moins 2 heures. |
Oubli de dose
Si l'oubli de la dose d'acalabrutinib remonte à plus de 3 heures, demander au patient de prendre la dose suivante à l'heure habituelle. Le patient ne doit pas prendre de dose double d'acalabrutinib pour compenser la dose oubliée.
Populations particulières
Sujets âgés
Aucun ajustement posologique n'est requis pour les patients âgés (≥65 ans) (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
Aucune étude clinique spécifique n'a été conduite chez des patients atteints d'insuffisance rénale. Des patients atteints d'insuffisance rénale légère ou modérée ont été traités dans les études cliniques avec acalabrutinib. Aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance rénale légère ou modérée (clairance de la créatinine supérieure à 30 mL/min). L'hydratation doit être maintenue et les taux de créatinine sériques surveillés périodiquement. Acalabrutinib ne doit être administré à des patients atteints d'insuffisance rénale sévère (clairance de la créatinine < 30 mL/min) que si le bénéfice l'emporte sur le risque et les patients doivent être surveillés attentivement à la recherche d'éventuels signes de toxicité. Il n'existe pas de données chez les patients atteints d'insuffisance rénale sévère ou dialysés (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucun ajustement posologique n'est recommandé chez les patients atteints d'insuffisance hépatique légère ou modérée (Child-Pugh A, Child-Pugh B, ou bilirubine totale comprise entre 1,5 et 3 fois la limite supérieure de la normale [LSN] avec ou sans élévation d'ASAT). Il n'est pas recommandé d'utiliser acalabrutinib chez les patients atteints d'insuffisance hépatique sévère (Child-Pugh C ou bilirubine totale >3 fois LSN avec ou sans élévation d'ASAT) (voir rubrique Propriétés pharmacocinétiques).
Maladie cardiaque sévère
Les patients atteints d'une maladie cardiovasculaire sévère ont été exclus des études cliniques sur acalabrutinib.
Population pédiatrique
La sécurité et l'efficacité d'acalabrutinib chez les enfants et les adolescents âgés de 0 à 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Acalabrutinib est destiné à être utilisé par voie orale. Les gélules doivent être avalées entières avec de l'eau à peu près à la même heure chaque jour, avec ou sans nourriture (voir rubrique Propriétés pharmacocinétiques). Les gélules ne doivent pas être mâchées, dissoutes ou ouvertes.
Durée de conservation :
3 ans.
Précautions particulières de conservation :
Ne pas conserver à une température supérieure à 30°C.
Sans objet.
Il n'existe aucun traitement spécifique en cas de surdosage en acalabrutinib et les symptômes du surdosage n'ont pas été établis. En cas de surdosage, les patients doivent être surveillés attentivement à la recherche de signes ou de symptômes d'effets indésirables et un traitement symptomatique approprié doit être mis en place.
Classe pharmacothérapeutique : Antinéoplasiques, inhibiteurs des protéines kinases, code ATC : L01XE5.
Mécanisme d'action
L'acalabrutinib est un inhibiteur sélectif de la tyrosine kinase de Bruton (BTK). La BTK est une molécule de signalisation des voies des récepteurs antigéniques des cellules B (BCR) et des récepteurs de cytokines. La signalisation induite par la BTK stimule la survie et la prolifération des lymphocytes B et est essentielle à l'adhésion, au transport et au chimiotactisme de ces cellules.
L'acalabrutinib et son métabolite actif, l'ACP-5862, forment une liaison covalente avec un résidu cystéine au niveau du site actif de la BTK, entraînant ainsi l'inactivation irréversible de la BTK avec des interactions hors cible minimes.
Effets pharmacodynamiques
Chez des patients atteints d'hémopathies malignes B et ayant reçu 100 mg deux fois par jour, une occupation médiane à l'état d'équilibre de la BTK de ≥95 % dans le sang périphérique a été maintenue sur 12 heures, entraînant l'inactivation de la BTK pendant l'intégralité de l'intervalle recommandé entre deux prises.
Électrophysiologie cardiaque
L'effet de l'acalabrutinib sur l'intervalle QTc a été évalué chez 46 sujets sains, de sexe masculin et féminin, dans le cadre d'une étude spécifique de l'intervalle QT randomisée en double aveugle, contrôlée versus placebo et témoin positif. À une dose suprathérapeutique correspondant à 4 fois la dose maximale recommandée, acalabrutinib n'a pas allongé l'intervalle QT/QTc dans une mesure cliniquement pertinente (soit pas de façon ≥ 10 ms) (voir rubriques Mises en garde et précautions d'emploi, Effets indésirables et Données de sécurité précliniques).
Efficacité et sécurité cliniques
Patients atteints d'une LLC non précédemment traités
La tolérance et l'efficacité d'acalabrutinib dans la LLC non précédemment traitée ont été évaluées dans une étude de phase III randomisée, multicentrique, en ouvert (ELEVATE-TN) chez 535 patients. Les patients ont reçu l'association acalabrutinib plus obinutuzumab, acalabrutinib en monothérapie, ou l'association obinutuzumab plus chlorambucil. Des patients âgés de 65 ans ou plus ou entre 18 et 65 ans avec des maladies coexistantes ont été inclus dans l'essai ELEVATE-TN, et 27,9 % des patients avaient une clairance de la créatinine (ClCr) < 60 mL/min. Parmi les patients âgés de < 65 ans,16,1 % avaient un score CIRS-G médian de 8. Dans cet essai, les patients pouvaient recevoir des antithrombotiques. Les patients qui nécessitaient une anticoagulation par warfarine ou antivitamines K équivalents ont été exclus.
Les patients ont été randomisés selon un ratio 1 : 1 : 1 dans 3 bras afin de recevoir :
· L'association acalabrutinib plus obinutuzumab (acalabrutinib +G) : 100 mg d'acalabrutinib ont été administrés deux fois par jour à partir du Jour 1 du Cycle 1 jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable. L'obinutuzumab a été administré à partir du Jour 1 du Cycle 2 pendant un maximum de 6 cycles de traitement. 1 000 mg d'obinutuzumab ont été administrés les Jours 1 et 2 (100 mg le Jour 1 et 900 mg le Jour 2), 8 et 15 du Cycle 2 puis 1 000 mg ont été administrés le Jour 1 des Cycles 3 à 7. Chaque cycle durait 28 jours.
· Acalabrutinib en monothérapie : 100 mg d'acalabrutinib ont été administrés deux fois par jour jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
· L'association obinutuzumab plus chlorambucil (GClb) : l'obinutuzumab et le chlorambucil ont été administrés pendant un maximum de 6 cycles de traitement. 1 000 mg d'obinutuzumab ont été administrés les Jours 1 et 2 (100 mg le Jour 1 et 900 mg le Jour 2), 8 et 15 du Cycle 1 puis 1 000 mg ont été administrés le Jour 1 des Cycles 2 à 6. Le chlorambucil a été administré à raison de 0,5 mg/kg les Jours 1 et 15 des Cycles 1 à 6. Chaque cycle durait 28 jours.
Les patients ont été stratifiés selon le statut mutationnel pour la délétion 17p (présence versus absence), l'indice de performance ECOG (0 ou 1 versus 2) et la région géographique (Amérique du Nord et Europe occidentale versus Autre). Après une progression confirmée de la maladie, 45 patients randomisés dans le bras GClb sont passés à acalabrutinib en monothérapie. Le Tableau 5 résume les données démographiques et les caractéristiques de la maladie à l'inclusion de la population de l'étude.
Tableau 5. Caractéristiques à l'inclusion des patients atteints d'une LLC non précédemment traités (étude ELEVATE-TN)
| Caractéristique | Acalabrutinib plus obinutuzumab N = 179 | Acalabrutinib en monothérapie N = 179 | Obinutuzumab plus chlorambucil N = 177 |
| Âge, années ; médiane (min-max) | 70 (41-88) | 70 (44-87) | 71 (46-91) |
| Hommes ; % | 62 | 62 | 59,9 |
| Caucasiens ; % | 91,6 | 95 | 93,2 |
| Indice de performance ECOG 0-1 ; % | 94,4 | 92,2 | 94,4 |
| Temps médian écoulé depuis le diagnostic (mois) | 30,5 | 24,4 | 30,7 |
| Masse tumorale volumineuse avec ganglions ≥ 5 cm ; % | 25,7 | 38 | 31,1 |
| Catégorie cytogénétique/FISH ; % Délétion 17p Délétion 11q Mutation de TP53 IGHV non muté Caryotype complexe (≥3 anomalies) | 9,5 17,3 11,7 57,5 16,2 | 8,9 17,3 10,6 66,5 17,3 | 9 18,6 11,9 65,5 18,1 |
| Stade Rai ; % | | | |
| 0 I II III IV | 1,7 30,2 20,1 26,8 21,2 | 0 26,8 24,6 27,9 20,7 | 0,6 28,2 27,1 22,6 21,5 |
Le critère d'évaluation principal était la survie sans progression (SSP) dans le bras acalabrutinib +G versus le bras GClb évaluée par un comité de revue indépendant (Independent Review Committee, IRC) d'après les critères IWCLL (International Workshop on Chronic Lymphocytic Leukaemia) 2008 avec incorporation de la clarification pour la lymphocytose liée au traitement (Cheson 2012). Avec un suivi médian de 28,3 mois, la SSP évaluée par un IRC montrait une réduction statistiquement significative de 90 % du risque de progression de la maladie ou de décès pour les patients atteints d'une LLC non précédemment traités dans le bras acalabrutinib +G versus le bras GClb. Les résultats d'efficacité sont présentés dans le Tableau 6. Les courbes de Kaplan-Meier pour la SSP sont présentées sur la Figure 1.
Tableau 6. Résultats d'efficacité chez les patients atteints d'une LLC (étude ELEVATE-TN)Temps écoulé depuis la randomisation (mois)
| | Acalabrutinib plus obinutuzumab N = 179 | Acalabrutinib en monothérapie N = 179 | Obinutuzumab plus chlorambucil N = 177 |
| Survie sans progression* | |||
| Nombre d'événements (%) | 14 (7,8 %) | 26 (14,5 %) | 93 (52,5 %) |
| PM, n (%) | 9 (5 %) | 20 (11,2 %) | 82 (46,3 %) |
| Événements de décès (%) | 5 (2,8 %) | 6 (3,4 %) | 11 (6,2 %) |
| Médiane (IC 95 %), mois | NA | NA (34,2 ; NA) | 22,6 (20,2 ; 27,6) |
| HR† (IC 95 %) | 0,10 (0,06 ; 0,17) | 0,20 (0,13 ; 0,30) | - |
| Valeur de p | <0,0001 | <0,0001 | - |
| Estimation à 24 mois, % (IC 95 %) | 92,7 (87,4 ; 95,8) | 87,3 (80,9 ; 91,7) | 46,7 (38,5 ; 54,6) |
| Survie globalea | | | |
| Événements de décès (%) | 9 (5 %) | 11 (6,1 %) | 17 (9,6 %) |
| HR (IC 95 %) † | 0,47 (0 21 ; 1,06) | 0,60 (0 28 ; 1,27) | - |
| Meilleur taux de réponse globale* (RC + RCi + RPn + RP) | |||
| TRG, n (%) (IC 95 %) | 168 (93,9) (89,3 ; 96,5) | 153 (85,5) (79,6 ; 89,9) | 139 (78,5) (71,9 ; 83,9) |
| Valeur de p | <0,0001 | 0,0763 | - |
| RC, n (%) | 23 (12,8) | 1 (0,6) | 8 (4,5) |
| RCi, n (%) | 1 (0,6) | 0 | 0 |
| RPn, n (%) | 1 (0,6 %) | 2 (1,1 %) | 3 (1,7 %) |
| RP, n (%) | 143 (79,9) | 150 (83,8) | 128 (72,3) |
IC = intervalle de confiance ; HR = hazard ratio ; NA = non atteinte ; RC = réponse complète ; RCi = réponse complète avec récupération incomplète de la numération sanguine ; RPn = réponse partielle nodulaire ; RP = réponse partielle
*D'après l'évaluation de l'IRC
†D'après un modèle à risques proportionnels de Cox stratifié
a La médiane de SG n'a été atteinte dans aucun des bras.
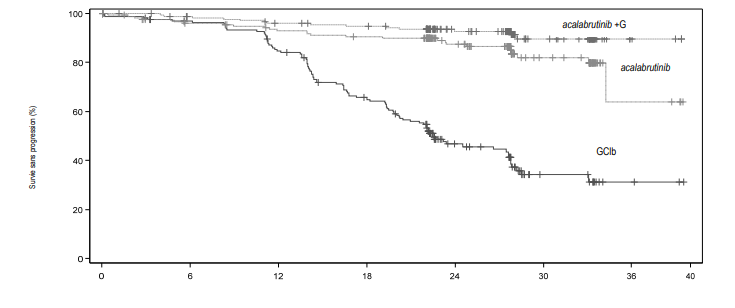
![]() Figure
1. Courbe de Kaplan-Meier de la SSP évaluée par un IRC chez les
patients atteints d'une LLC (Population ITT) (étude ELEVATE-TN)
Figure
1. Courbe de Kaplan-Meier de la SSP évaluée par un IRC chez les
patients atteints d'une LLC (Population ITT) (étude ELEVATE-TN)
| Nombre de patients à risque | ||||||||||||||
| Mois | 0 | 3 | 6 | 9 | 12 | 15 | 18 | 21 | 24 | 27 | 30 | 33 | 36 | 39 |
| Acalabrutinib | 179 | 166 | 161 | 157 | 153 | 150 | 148 | 147 | 103 | 94 | 43 | 40 | 4 | 3 |
| Acalabrutinib +G | 179 | 176 | 170 | 168 | 163 | 160 | 159 | 155 | 109 | 104 | 46 | 41 | 4 | 2 |
| GClb | 177 | 162 | 157 | 151 | 136 | 113 | 102 | 86 | 46 | 41 | 13 | 13 | 3 | 2 |
Les résultats de SSP pour acalabrutinib avec ou sans obinutuzumab étaient constants dans les différents sous-groupes, y compris pour les caractéristiques à haut risque. Dans la population de patients atteints d'une LLC à haut risque (délétion 17p, délétion 11q, mutation de TP53 ou IGHV non muté), les HR pour la SSP d'acalabrutinib avec ou sans obinutuzumab versus l'association obinutuzumab plus chlorambucil étaient de 0,08 (IC 95 % [0,04 ; 0,15]) et 0,13 (IC 95 % [0,08 ; 0,21]), respectivement.
Tableau 7. Analyse en sous-groupes de la SSP (étude ELEVATE-TN)
| | Acalabrutinib en monothérapie | Acalabrutinib +G | ||||
| N | Hazard Ratio | IC 95 % | N | Hazard Ratio | IC 95 % | |
| Tous les patients | 179 | 0,20 | (0,13 ; 0,30) | 179 | 0,10 | (0,06 ; 0,17) |
| Del 17p | | | | | | |
| Oui | 19 | 0,20 | (0,06 ; 0,64) | 21 | 0,13 | (0,04 ; 0,46) |
| Non | 160 | 0,20 | (0,12 ; 0,31) | 158 | 0,09 | (0,05 ; 0,17) |
| Mutation | | | | | | |
| de TP53 | | | | | | |
| Oui | 19 | 0,15 | (0,05 ; 0,46) | 21 | 0,04 | (0,01 ; 0,22) |
| Non | 160 | 0,20 | (0,12 ; 0,32) | 158 | 0,11 | (0,06 ; 0,20) |
| Del 17p | | | | | | |
| et/ou | | | | | | |
| mutation | | | | | | |
| de TP53 | | | | | | |
| Oui | 23 | 0,10 | (0 03 ; 0,34) | 25 | (0 03 ; 0,34) | (0 09 ; 0,48) |
| Non | 156 | 0,10 | (0 05 ; 0,18) | 154 | (0 05 ; 0,18) | (0 21 ; 0,61) |
| Mutation | | | | | | |
| d'IGHV | | | | | | |
| Muté | 58 | 0,69 | (0,31 ; 1,56) | 74 | 0,15 | (0,04 ; 0,52) |
| Non muté | 119 | 0,11 | (0,07 ; 0,19) | 103 | 0,08 | (0,04 ; 0,16) |
| Del 11q | | | | | | |
| Oui | 31 | 0,07 | (0,02 ; 0,22) | 31 | 0,09 | (0,03 ; 0,26) |
| Non | 148 | 0,26 | (0,16 ; 0,41) | 148 | 0,10 | (0,05 ; 0,20) |
| Caryotype | | | | | | |
| complexe | | | | | | |
| Oui | 31 | 0,10 | (0,03 ; 0,33) | 29 | 0,09 | (0,03 ; 0,29) |
| Non | 117 | 0,27 | (0,16 ; 0,46) | 126 | 0,11 | (0,05 ; 0,21) |
Patients atteints d'une LLC ayant reçu au moins un traitement antérieur
La tolérance et l'efficacité d'acalabrutinib dans la LLC en rechute ou réfractaire ont été évaluées dans une étude de phase III randomisée, multicentrique, en ouvert (ASCEND) chez 310 patients ayant reçu au moins un traitement antérieur, hors inhibiteurs de BCL-2 ou inhibiteurs du récepteur des cellules B. Les patients ont reçu soit acalabrutinib en monothérapie soit, au choix de l'investigateur, l'association idélalisib plus rituximab ou l'association bendamustine plus rituximab. Dans cet essai, les patients pouvaient recevoir des antithrombotiques. Les patients qui nécessitaient une anticoagulation par warfarine ou antivitamines K équivalents ont été exclus.
Les patients ont été randomisés selon un ratio 1 : 1 afin de recevoir :
· Acalabrutinib à raison de 100 mg deux fois par jour jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable, ou
· Au choix de l'investigateur :
o L'idélalisib à raison de 150 mg deux fois par jour jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable en association avec ≤ 8 perfusions de rituximab (375 mg/m2/500 mg/m2) le Jour 1 de chaque cycle de 28 jours pendant un maximum de 6 cycles
o La bendamustine à raison de 70 mg/m2 (Jours 1 et 2 de chaque cycle de 28 jours) en association avec le rituximab (375 mg/m2/500 mg/m2) le Jour 1 de chaque cycle de 28 jours pendant un maximum de 6 cycles
Les patients ont été stratifiés selon le statut mutationnel pour la délétion 17p (présence versus absence), l'indice de performance ECOG (0 ou 1 versus 2) et le nombre de traitements antérieurs (1 à 3 versus ≥4). Après une progression confirmée de la maladie, 35 patients randomisés pour recevoir, au choix de l'investigateur, l'association idélalisib plus rituximab ou l'association bendamustine plus rituximab sont passés à acalabrutinib. Le Tableau 8 résume les données démographiques et les caractéristiques de la maladie à l'inclusion de la population de l'étude.
Tableau 8. Caractéristiques à l'inclusion des patients atteints d'une LLC (Étude ASCEND)
| Caractéristique | Acalabrutinib en monothérapie N = 155 | Au choix de l'investigateur, idélalisib + rituximab ou bendamustine + rituximab N = 155 | |||||
| Âge, années ; médiane (min-max) | 68 (32-89) | 67 (34-90) | |||||
| Hommes ; % | 69,7 | 64,5 | |||||
| Caucasiens ; % | 93,5 | 91,0 | |||||
| Indice de performance ECOG ; % | | | |||||
| 0 | 37,4 | 35,5 | |||||
| 1 | 50,3 | 51,0 | |||||
| 2 | 12,3 | 13,5 | |||||
| Temps (mois) | médian | écoulé | depuis | le | diagnostic | 85,3 | 79,0 |
| Masse tumorale volumineuse avec ganglions ≥5 cm ; % | 49,0 | 48,4 | |||||
| Nombre médian de traitements antérieurs pour la LLC (min-max) | 1 (1-8) | 2 (1-10) | |||||
| Nombre de traitements antérieurs pour la LLC ; % 1 2 3 ≥4 | 52,9 25,8 11,0 10,3 | 43,2 29,7 15,5 11,6 | |||||
| Catégorie cytogénétique/FISH ; % | | | |||||
| Délétion 17p | 18,1 | 13,5 | |||||
| Délétion 11q | 25,2 | 28,4 | |||||
| Mutation de TP53 | 25,2 | 21,9 | |||||
| IGHV non muté | 76,1 | 80,6 | |||||
| Caryotype complexe (≥3 anomalies) | 32,3 | 29,7 | |||||
| Stade Rai ; % | | | |||||
| 0 | 1,3 | 2,6 | |||||
| I | 25,2 | 20,6 | |||||
| II | 31,6 | 34,8 | |||||
| III | 13,5 | 11,6 | |||||
| IV | 28,4 | 29,7 | |||||
Le critère d'évaluation principal était la SSP évaluée par un IRC d'après les critères IWCLL 2008 avec incorporation de la clarification pour la lymphocytose liée au traitement (Cheson 2012). Avec un suivi médian de 16,1 mois, la SSP montrait une réduction statistiquement significative de 69 % du risque de décès ou de progression pour les patients du bras acalabrutinib. Les résultats d'efficacité sont présentés dans le Tableau 9. La courbe de Kaplan-Meier pour la SSP est présentée sur la Figure 2.
Tableau 9. Résultats d'efficacité chez les patients atteints d'une LLC (étude ASCEND)
| | Acalabrutinib en monothérapie N = 155 | Au choix de l'investigateur, idélalisib + rituximab ou bendamustine + rituximab N = 155 |
| Survie sans progression* | ||
| Nombre d'événements (%) | 27 (17,4) | 68 (43,9) |
| PM, n (%) | 19 (12,3) | 59 (38,1) |
| Événements de décès (%) | 8 (5,2) | 9 (5,8) |
| Médiane (IC 95 %), mois | NA | 16,5 (14,0 ; 17,1) |
| HR† (IC 95 %) | 0,31 (0,20 ; 0,49) | |
| Valeur de p | <0,0001 | |
| Estimation à 15 mois, % (IC 95 %) | 82,6 (75,0 ; 88,1) | 54,9 (45,4 ; 63,5) |
| Survie globalea | | |
| Événements de décès (%) | 15 (9,7 %) | 18 (11,6 %) |
| HR (IC 95 %) † | 0,84 (0 42 ;1,66) | - |
| Meilleur taux de réponse globale* (RC + RCi + RPn + RP)** | ||
| TRG, n (%) (IC 95 %) | 126 (81,3) (74,4 ; 86,6) | 117 (75,5) (68,1 ; 81,6) |
| Valeur de p | 0,2248 | - |
| RC, n (%) | 0 | 2 (1,3) |
| RP, n (%) | 126 (81,3) | 115 (74,2) |
| Durée de la réponse (DR) | ||
| Médiane (IC 95 %), mois | NA | 13,6 (11,9 ; NA) |
IC = intervalle de confiance ; HR = hazard ratio ; NA = non atteinte ; RC = réponse complète ; RCi = réponse complète avec récupération incomplète de la numération sanguine ; RPn = réponse partielle nodulaire ; RP = réponse partielle
*D'après l'évaluation de l'IRC
a La médiane de SG n'a été atteinte dans aucun des bras. P < 0,6089 pour la SG.
**La RCi et la RPn ont des valeurs de 0
†D'après un modèle à risques proportionnels de Cox stratifié
Figure 2. Courbe de Kaplan-Meier de la SSP évaluée par un IRC chez les patients atteints d'une LLC (Population ITT) (étude ASCEND)
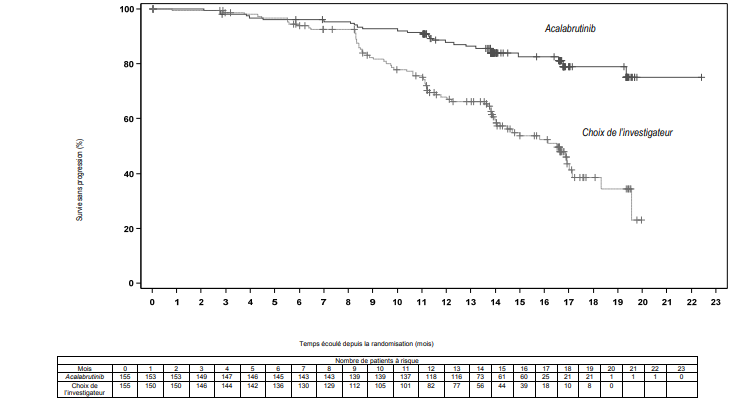
Les résultats de SSP pour acalabrutinib étaient constants dans les différents sous-groupes, y compris pour les caractéristiques à haut risque. Dans la population de patients atteints d'une LLC à haut risque (délétion 17p, délétion 11q, mutation de TP53 et IGHV non muté), le HR pour la SSP était de 0,27 (IC 95 % [0,17 ; 0,44]).
Tableau 10. Analyse en sous-groupes de la SSP (étude ASCEND)
| | Acalabrutinib en monothérapie | ||
| N | Hazard Ratio | IC 95 % | |
| Tous les patients | 155 | 0,30 | (0,19 ; 0,48) |
| Del 17p Oui Non | 28 127 | 0,21 0,33 | (0,07 ; 0,68) (0,21 ; 0,54) |
| Mutation de TP53 Oui Non | 39 113 | 0,24 0,33 | (0,11 ; 0,56) (0,20 ; 0,57) |
| Del 17p ou mutation de TP53 Oui Non | 45 108 | 0,21 0,36 | (0 09 ; 0,48) (0 21 ; 0,61) |
| Mutation d'IGHV Muté Non muté | 33 118 | 0,32 0,32 | (0,11 ; 0,94) (0,19 ; 0,52) |
| Del 11q Oui Non | 39 116 | 0,28 0,31 | (0,11 ; 0,70) (0,19 ; 0,53) |
| Caryotype complexe Oui Non | 50 97 | 0,32 0,23 | (0,16 ; 0,63) (0,12 ; 0,44) |
La pharmacocinétique (PK) de l'acalabrutinib et de son métabolite actif, l'ACP-5862, a été étudiée chez des sujets sains et des patients atteints d'hémopathies malignes B. La PK de l'acalabrutinib est proportionnelle à la dose, et l'acalabrutinib ainsi que l'ACP-5862 présentent une PK presque linéaire sur un intervalle de doses de 75 à 250 mg. La modélisation PK de population suggère que la PK de l'acalabrutinib et de l'ACP-5862 ne diffère pas significativement chez les patients atteints de différentes hémopathies malignes B. À la dose recommandée de 100 mg deux fois par jour chez des patients atteints d'hémopathies malignes B (y compris LLC), les moyennes géométriques de l'aire sous la courbe quotidienne de la concentration plasmatique en fonction du temps (ASC24h) et de la concentration plasmatique maximale (Cmax) à l'état d'équilibre pour l'acalabrutinib étaient de 1 893 ng•h/mL et 466 ng/mL, respectivement, et pour l'ACP-5862, ces valeurs étaient de 4 091 ng•h/mL et 420 ng/mL, respectivement.
Absorption
Le temps médian nécessaire pour atteindre les concentrations plasmatiques maximales (Tmax) était de 0,75 heure pour l'acalabrutinib, et de 1,0 heure pour l'ACP-5862. La biodisponibilité absolue d'acalabrutinib était de 25 %.
Effet de la nourriture sur l'acalabrutinib
Chez des sujets sains, l'administration d'une dose unique de 75 mg d'acalabrutinib avec un repas riche en lipides et en calories (environ 918 calories, 59 grammes de glucides, 59 grammes de lipides et 39 grammes de protéines) n'a pas affecté l'ASC moyenne par rapport à l'administration à jeun. La Cmax résultante a diminué de 69 % et le Tmax a été allongé de 1 à 2 heures.
Distribution
La liaison réversible aux protéines plasmatiques humaines était de 97,5 % pour l'acalabrutinib et de 98,6 % pour l'ACP-5862. In vitro, le ratio moyen sang/plasma était de 0,8 pour l'acalabrutinib et de 0,7 pour l'ACP-5862. Le volume de distribution moyen à l'état d'équilibre (Vee) était d'environ 34 L pour l'acalabrutinib.
Biotransformation / métabolisme
In vitro, l'acalabrutinib est principalement métabolisé par les enzymes CYP3A, et dans une moindre mesure par conjugaison au glutathion et hydrolyse de la fonction amide. L'ACP-5862 est le principal métabolite mis en évidence dans le plasma, celui-ci ayant ensuite été principalement métabolisé par oxydation médiée par le CYP3A, et la moyenne géométrique de son exposition (ASC) était de 2 à 3 fois plus élevée que celle de l'acalabrutinib. L'ACP-5862 est environ 50 % moins puissant que l'acalabrutinib en termes d'inhibition de la BTK.
Les études in vitro indiquent que l'acalabrutinib n'inhibe pas le CYP1A2, le CYP2B6, le CYP2C8, le CYP2C9, le CYP2C19, le CYP2D6, le CYP3A4/5, l'UGT1A1 ni l'UGT2B7 aux concentrations cliniquement pertinentes et qu'il ne devrait pas affecter la clairance des substrats de ces CYP.
Les études in vitro indiquent que l'ACP-5862 n'inhibe pas le CYP1A2, le CYP2B6, le CYP2C8, le CYP2C9, le CYP2C19, le CYP2D6, le CYP3A4/5, l'UGT1A1 ni l'UGT2B7 aux concentrations cliniquement pertinentes et qu'il ne devrait pas affecter la clairance des substrats de ces CYP.
Interactions avec les protéines de transport
L'acalabrutinib peut inhiber la BCRP intestinale (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions), tandis que l'ACP-5862 peut inhiber la MATE1 (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions) aux concentrations cliniquement pertinentes. L'acalabrutinib n'inhibe pas la MATE1, tandis que l'ACP-5862 n'inhibe pas la BCRP aux concentrations cliniquement pertinentes.
Les études in vitro indiquent que l'acalabrutinib et l'ACP-5862 sont des substrats de la P-glycoprotéine (P-gp) et de la protéine de résistance du cancer du sein (breast cancer resistance protein, BCRP). L'acalabrutinib peut augmenter l'exposition aux substrats de la P-gp et de la BCRP co-administrés par inhibition de la P-gp ou de la BCRP intestinale (voir rubrique Posologie et mode d'administration). La co-administration avec un inhibiteur de l'OATP1B1/1B3 (600 mg de rifampicine en dose unique) a multiplié la Cmax et l'ASC de l'acalabrutinib respectivement par 1,2 et 1,4 (N = 24, sujets sains), ce qui n'est pas cliniquement pertinent. L'ACP-5862 n'est pas un substrat de l'OATP1B1 ni de l'OATP1B3.
L'acalabrutinib et l'ACP-5862 n'inhibent pas la P-gp, l'OAT1, l'OAT3, l'OCT2, l'OATP1B1, l'OATP1B3 et la MATE2-K aux concentrations cliniquement pertinentes.
Élimination
Après une dose unique par voie orale de 100 mg d'acalabrutinib, la demi-vie d'élimination terminale (t1/2) médiane de l'acalabrutinib était de 0,9 (min-max : 0,6 à 2,8) heure. La t1/2 médiane du métabolite actif, l'ACP-5862, était de 6,9 (min-max : 2,7 à 9,1) heures.
La clairance orale apparente (CL/F) moyenne était de 70 L/h pour l'acalabrutinib et de 13 L/h pour l'ACP-5862, avec une PK similaire entre les patients et les sujets sains d'après une analyse PK de population.
Après l'administration d'une dose unique de 100 mg d'acalabrutinib radiomarqué au [14C] chez des sujets sains, 84 % de la dose a été retrouvée dans les fèces et 12 % dans les urines, moins de 2 % de la dose ayant été excrétée sous forme inchangée.
Populations particulières
D'après une analyse PK de population, l'âge (> 18 ans), le sexe, l'origine ethnique (Caucasiens, Afro- Américains) et le poids corporel n'ont pas eu d'effets cliniquement significatifs sur la PK de l'acalabrutinib et de son métabolite actif, l'ACP-5862.
Population pédiatrique
Aucune étude pharmacocinétique n'a été conduite avec acalabrutinib chez les patients de moins de 18 ans.
Insuffisance rénale
L'élimination rénale de l'acalabrutinib est minime. Aucune étude pharmacocinétique n'a été conduite chez des patients atteints d'insuffisance rénale.
D'après une analyse PK de population, aucune différence cliniquement pertinente n'a été observée au niveau de la PK chez 433 patients atteints d'insuffisance rénale légère (DFGe compris entre 60 et 89 mL/min/1,73 m2 d'après l'équation MDRD), 110 patients atteints d'insuffisance rénale modérée (DFGe compris entre 30 et 59 mL/min/1,73 m2) versus 204 patients présentant une fonction rénale normale (DFGe supérieur ou égal à 90 mL/min/1,73 m2). La pharmacocinétique de l'acalabrutinib n'a pas été caractérisée chez les patients atteints d'insuffisance rénale sévère (DFGe inférieur à 29 mL/min/1,73 m2) ou d'insuffisance rénale nécessitant une dialyse. Les patients présentant des taux de créatinine correspondant à plus de 2,5 fois la LSN de l'établissement n'ont pas été inclus dans les essais cliniques (voir rubrique Posologie et mode d'administration).
Insuffisance hépatique
L'acalabrutinib est métabolisé dans le foie. Dans des études spécifiques de l'insuffisance hépatique, par rapport aux sujets présentant une fonction hépatique normale (n = 6), l'exposition (ASC) à l'acalabrutinib a été multipliée par 1,9 ; 1,5 et 5,3 chez les sujets atteints d'insuffisance hépatique légère (n = 6) (Child-Pugh A), modérée (n = 6) (Child-Pugh B) et sévère (n = 8) (Child-Pugh C), respectivement. D'après une analyse PK de population, aucune différence cliniquement pertinente n'a été observée entre les sujets atteints d'insuffisance hépatique légère (n = 79) ou modérée (n = 6) (bilirubine totale comprise entre 1,5 et 3 fois la LSN avec ou sans élévation d'ASAT) et les sujets présentant une fonction hépatique normale (n = 651) (taux de bilirubine totale et d'ASAT inférieurs à la LSN) (voir rubrique Posologie et mode d'administration).
Acalabrutinib n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Cependant, pendant le traitement par acalabrutinib, de la fatigue et des étourdissements ont été rapportés et il doit être conseillé aux patients qui présentent ces symptômes de ne pas conduire de véhicules et de ne pas utiliser de machines jusqu'à la disparition des symptômes.
Carcinogénicité
Aucune étude de carcinogénicité n'a été conduite avec l'acalabrutinib.
Génotoxicité/mutagénicité
L'acalabrutinib s'est révélé non mutagène dans un test de mutation réverse sur bactéries, un test d'aberrations chromosomiques in vitro et un test in vivo du micronoyau dans la moelle osseuse chez la souris.
Toxicité en administration répétée
Chez le rat, des anomalies microscopiques d'intensité minime à légère ont été observées au niveau du pancréas (hémorragie/pigment/inflammation/fibrose au niveau des îlots) à tous les niveaux de dose. Des anomalies non indésirables d'intensité minime à légère ont été observées au niveau des reins (basophilie tubulaire, régénération tubulaire et inflammation) dans des études d'une durée allant jusqu'à 6 mois, avec une dose sans effet toxique observable (No Observed Adverse Effect Level, NOAEL) de 30 mg/kg/jour chez le rat femelle et de 100 mg/kg/jour chez le mâle. Les expositions moyennes (ASC) à la NOAEL chez les rats mâles et femelles correspondent respectivement à 2,5 et 1 fois l'exposition clinique à la dose recommandée de 100 mg deux fois par jour. La plus faible dose entraînant un effet toxique observable (Lowest Observed Adverse Effect Level, LOAEL) à laquelle des anomalies rénales (dégénérescence tubulaire modérée) et hépatiques (nécrose hépatocytaire individuelle) réversibles ont été observées dans l'étude de toxicité chronique conduite chez le rat était de 100 mg/kg/jour et a donné une marge d'exposition correspondant à 4,2 fois l'exposition clinique à la dose recommandée de 100 mg deux fois par jour. Des toxicités cardiaques (hémorragie, inflammation et nécrose au niveau du myocarde) ont été observées chez les rats morts pendant l'étude à des doses supérieures à la dose maximale tolérée (maximum tolerated dose, MTD). L'exposition chez les rats présentant des anomalies cardiaques correspondait à au moins 6,8 fois l'exposition humaine à la dose clinique de 100 mg deux fois par jour. La réversibilité des anomalies cardiaques n'a pas pu être évaluée car ces anomalies ont uniquement été observées aux doses supérieures à la MTD.
Toxicité sur la reproduction
Aucun effet sur la fertilité n'a été observé chez les rats mâles ou femelles à des expositions correspondant à 10 ou 9 fois l'exposition basée sur l'ASC chez l'être humain à la dose recommandée, respectivement.
Aucun effet n'a été observé sur le développement embryo-foetal et la survie chez les rates gravides à des expositions correspondant à environ 9 fois l'ASC observée chez les patients à la dose recommandée de 100 mg deux fois par jour. La présence de l'acalabrutinib et de son métabolite actif a été confirmée dans le plasma foetal des rats. L'acalabrutinib et son métabolite actif étaient présents dans le lait de rates allaitantes.
Dans une étude embryo-foetale conduite chez des lapines gravides, une diminution du poids des foetus et un retard d'ossification ont été observés aux niveaux d'exposition ayant produit une toxicité maternelle, correspondant à 2,4 fois les niveaux d'exposition chez l'être humain à la dose recommandée.
Dans une étude de la reproduction conduite chez le rat, une dystocie (mise à bas prolongée/difficile) a été observée à des expositions correspondant à >2,3 fois l'exposition clinique à 100 mg deux fois par jour.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Médicament soumis à prescription hospitalière.
Médicament à prescription réservée aux spécialistes en hématologie ou aux médecins compétents en maladie du sang
Médicament nécessitant une surveillance particulière pendant le traitement.
Gélule.
Gélule de taille 1 dotée d'un corps jaune et d'une coiffe bleue, portant l'inscription « ACA 100 mg » à l'encre noire.
Plaquettes en aluminium/aluminium de 8 gélules (avec symboles du soleil/de la lune). Boîtes de 56 (7 x 8) gélules.